固有免疫在哮喘发病中的作用
2016/11/14
吴昌归
第四军医大学西京医院呼吸与危重症医学科,西安,710032
第四军医大学西京医院呼吸与危重症医学科,西安,710032
长期以来人们认为哮喘是一种免疫系统疾病,主要由Th2介导。Th2细胞通过产生和释放IL-4、IL-5、IL-9和IL-13等细胞因子,促进B细胞合成IgE,刺激Th2分化、诱导嗜酸性粒细胞、肥大细胞分化、发育、成熟和活化,引发气道高分泌和气道平滑肌异常收缩,在过敏性哮喘的发生发展中起关键作用。然而,近几年的研究表明,哮喘是一种异质性很强的复杂疾病,靶向Th2细胞因子的治疗策略仅使部分哮喘患者受益,提示除Th2细胞参与哮喘发病外,还有其他免疫活性细胞参与其中,尤其在非过敏性哮喘的发生发展中起一定作用。非过敏性哮喘主要与环境因素(如污染、臭氧、吸烟、汽车尾气、病毒感染、应激等)和营养失调(肥胖)有关,其发病独立于Th2机制,中性粒细胞等其他固有免疫细胞所起作用受到学界关注。本文综述近几年这一领域的研究成果,以飨读者。
一、过敏性哮喘发生的经典途径(适应性免疫——Th2途径)
在出生之初气道内没有抗原递呈细胞(主要是树突状细胞,DC),当感染和其他刺激损伤或激活气道上皮后,使之释放一系列趋化因子如CCL20、CCL19和CCL27,趋化DC由骨髓移向粘膜上皮和粘膜下组织,在此处DC逐渐成熟,获得摄取并处理过敏原的能力[1]。当过敏原进入气道后即被DC摄取,并处理成抗原肽,之后DC又归巢至区域淋巴结T细胞区,并通过MHC I或II类分子将有效抗原肽递呈给初始型T细胞表面受体(TCR),T细胞在接受抗原刺激后可向Th1或Th2分化,如果环境中有足够的IL-4,那么Th细胞就会向Th2亚群分化,Th2细胞部分移行至淋巴结的生发中心,与B细胞相互作用,促进其免疫球蛋白合成开关转换,由合成IgM转化为合成IgE,另一部分Th2细胞则移向气道粘膜,通过释放致敏性细胞因子,导致经典的Th2反应。
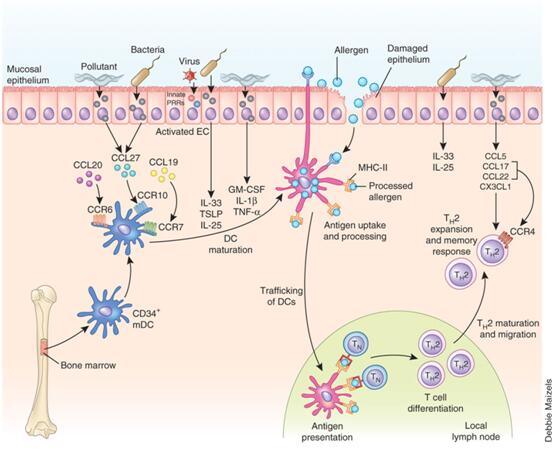
图1.过敏性哮喘的经典产生途径及其调节
二、固有免疫系统辅助参与过敏性哮喘的发生
在上述过敏性哮喘经典发病过程中,许多环节将受到主要来自于固有免疫系统的多种因素的影响和调节,如图(1)。
1.不成熟DC由骨髓迁移至气道粘膜过程的调节:作为固有免疫系统的重要成份,粘膜上皮细胞表面分布着大量的模式识别受体(PRRs,固有免疫系统的主要受体),TLRs是其中介导对来自细菌、病毒、真菌及其产物等配体反应的关键受体。如内毒素匹配TLR4,G+菌的脂蛋白与TLR2、6结合,病毒双链或单链RNA则分别结合TLR3、7/8,含CpG序列的细菌DNA对应于TLR9[2];其他PRR则对组织损伤产生的危险信号(DAMP),如游离ATP和尿酸等作出反应[3]。吸入过敏原的蛋白溶解活性、环境因素(如呼吸道病毒、空气污染等)对气道粘膜等组织产生损害,不仅增加过敏原透过粘膜屏障进入气道组织,还提供大量的危险信号(PAMP和DAMP),或由于伴随吸入的小剂量TLR配体,通过细胞表面的PRR激活上皮细胞,使之分泌一系列趋化物质,如:CCL20、CCL19、CCL27等,与DC表面的相应受体CCR6、CCR7和CCR10相互作用,不成熟DC便由骨髓移行至粘膜和粘膜下,在此逐渐发育成熟。
2.DC活化过程的调节:初移行至粘膜或粘膜下组织的DC摄取和加工抗原的能力很弱,必须活化并逐渐成熟才能完成适应性免疫过程。由于DC和上皮细胞表面的均表达TLR,上述危险信号直接作用于DC,或通过刺激气道粘膜上皮细胞合成和释放趋化因子(CCL17、CCL22)、细胞因子(IL-25、IL-33、GM-CSF和TSLP),使DC活化,其移动能力、摄取和加工抗原能力大大增强[4]。
3.DC成熟过程调节:DC一经成熟即失去其摄取和加工原能力,逐渐获得归巢区域引流淋巴结和提呈抗原能力。危险信号作用于DC使之上调趋化因子受体(归巢受体)、MHC I、MHC II、粘附分子和共刺激分子,指引其移行至淋巴结的T细胞区,并将抗原有效地提呈给T细胞。
4.Th2分化环境的形成:Th0细胞在接受MHC-抗原肽复合物刺激后可向Th1、Th2、Th9和Th17等亚型分化(图2)。而Th2分化必须有IL-4参与。由图1可知DC处于启动和调控获得性免疫反应的中心地位,将抗原递呈给T细胞并刺激其增殖,但DC不表达Th2分化的关键细胞因子——IL-4,那么,它从何而来(尤其是初始致敏个体)目前尚不清楚。有研究表明:固有淋巴样细胞II(ILC2s)、嗜酸性粒细胞、嗜碱性粒细胞、肥大细胞和NKT细胞等在IL-25、IL-33、GM-CSF和TSLP等上皮源性细胞因子作用下活化并释放Th2型细胞因子(IL-4等),为Th2分化提供了必要的环境。
1.不成熟DC由骨髓迁移至气道粘膜过程的调节:作为固有免疫系统的重要成份,粘膜上皮细胞表面分布着大量的模式识别受体(PRRs,固有免疫系统的主要受体),TLRs是其中介导对来自细菌、病毒、真菌及其产物等配体反应的关键受体。如内毒素匹配TLR4,G+菌的脂蛋白与TLR2、6结合,病毒双链或单链RNA则分别结合TLR3、7/8,含CpG序列的细菌DNA对应于TLR9[2];其他PRR则对组织损伤产生的危险信号(DAMP),如游离ATP和尿酸等作出反应[3]。吸入过敏原的蛋白溶解活性、环境因素(如呼吸道病毒、空气污染等)对气道粘膜等组织产生损害,不仅增加过敏原透过粘膜屏障进入气道组织,还提供大量的危险信号(PAMP和DAMP),或由于伴随吸入的小剂量TLR配体,通过细胞表面的PRR激活上皮细胞,使之分泌一系列趋化物质,如:CCL20、CCL19、CCL27等,与DC表面的相应受体CCR6、CCR7和CCR10相互作用,不成熟DC便由骨髓移行至粘膜和粘膜下,在此逐渐发育成熟。
2.DC活化过程的调节:初移行至粘膜或粘膜下组织的DC摄取和加工抗原的能力很弱,必须活化并逐渐成熟才能完成适应性免疫过程。由于DC和上皮细胞表面的均表达TLR,上述危险信号直接作用于DC,或通过刺激气道粘膜上皮细胞合成和释放趋化因子(CCL17、CCL22)、细胞因子(IL-25、IL-33、GM-CSF和TSLP),使DC活化,其移动能力、摄取和加工抗原能力大大增强[4]。
3.DC成熟过程调节:DC一经成熟即失去其摄取和加工原能力,逐渐获得归巢区域引流淋巴结和提呈抗原能力。危险信号作用于DC使之上调趋化因子受体(归巢受体)、MHC I、MHC II、粘附分子和共刺激分子,指引其移行至淋巴结的T细胞区,并将抗原有效地提呈给T细胞。
4.Th2分化环境的形成:Th0细胞在接受MHC-抗原肽复合物刺激后可向Th1、Th2、Th9和Th17等亚型分化(图2)。而Th2分化必须有IL-4参与。由图1可知DC处于启动和调控获得性免疫反应的中心地位,将抗原递呈给T细胞并刺激其增殖,但DC不表达Th2分化的关键细胞因子——IL-4,那么,它从何而来(尤其是初始致敏个体)目前尚不清楚。有研究表明:固有淋巴样细胞II(ILC2s)、嗜酸性粒细胞、嗜碱性粒细胞、肥大细胞和NKT细胞等在IL-25、IL-33、GM-CSF和TSLP等上皮源性细胞因子作用下活化并释放Th2型细胞因子(IL-4等),为Th2分化提供了必要的环境。
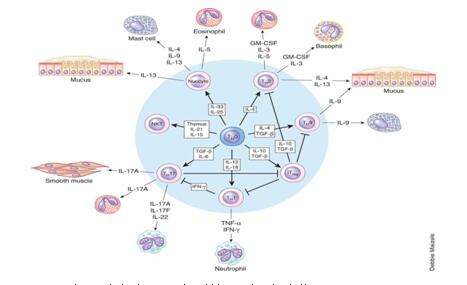
图2.涉及到哮喘不同表型的Th细胞分化
三、固有免疫在非适应性免疫哮喘发病中起主导作用
二十多年前就认为过敏性即Th2型哮喘是一种适应性免疫系统疾病,包括DC、肥大细胞、嗜酸性粒细胞、嗜碱性粒细胞和气道上皮细胞等固有免疫细胞作为辅助性效应细胞参与其中,但近来的研究和临床实践已越来越清楚地表明哮喘是一组具有很高异质性和复杂性的疾病,至少包括如过敏性和非过敏性在内的多种不同表型,单以Th2机制无法解释诸如非过敏性环境因素,如空气污染后涉及的固有免疫反应。在固有免疫中自然杀伤性T淋巴细胞(NKT)、固有淋巴样细胞2型(ILC2)和ILC3与哮喘关系研究较多。
一)iNKT细胞
NKT细胞具有NK细胞和T细胞特征,表达T细胞受体(TCR)和NK细胞受体(NKR-P1),前者识别由CD1a提呈的糖脂抗原,后者识别各种糖链。部分NKT所表达的TCR为半恒定TCR(iTCR),小鼠为Vα14 Jα18,而人类则为Vα24 Jα18。表达iTCR的NKT称iNKT,在固有免疫系统中发挥重要作用。在受体刺激后,它能较常规T细胞更迅速产生细胞因子,如IL-4、IFN-γ、IL-5、13和IL-17等,这一特点赋予其调节多种炎症性疾病的能力。
研究发现小鼠吸入曲霉菌提取物后在缺乏佐剂的情况下,非常有效地致敏小鼠并引发AHR,这种现象甚至发生在MHC II分子敲除后缺乏CD4+T淋巴细胞的小鼠模型上[5]。曲霉菌提取物中主要含鞘糖脂——asperamide B,与CD1a分子结合递呈给iNKT细胞,使之活化,分泌IL-4和IL-13。肺内活化的iNKT与巨噬细胞和上皮细胞相互作用,使它们分泌IL-33,激活肥大细胞、嗜酸性粒细胞、嗜碱性粒细胞、Th2细胞、iNKT和ILC2等细胞分泌IL-5和IL-13,直接或通过强化适应性免疫途径导致哮喘。其他含有糖脂的环境因子如屋尘螨、花粉等也能通过上述途径引发哮喘。此外,空气污染如臭氧能直接刺激小鼠iNKT分泌IL-17产生AHR[6]。
越来越多的实验研究尤其是哮喘小鼠研究表明,iNKT细胞在气道炎症和AHR的形成中起到重要作用。但由于功能性清除人iNKT的方法至今还不完善,因此它在人类哮喘发生中的作用均基于小鼠模型研究结果所推测。
二)ILC1
ILC1与NK细胞相关,均表达T-bet转录因子和IFN-γ;与NK不同处就是ILC1缺乏细胞毒作用,无穿孔素和颗粒酶。IL-12和IL-15使之活化(图3)。存在于结肠炎小鼠和克隆氏病患者肠道内,其与哮喘的关系相对于ILC2和ILC3来说远未明了,这主要由于研究方法的限制。推测ILC1像NK细胞一样参与哮喘患者或小鼠模型嗜酸性粒细胞气道炎——这一多数哮喘表型都能见到的特征性病理改变的形成。
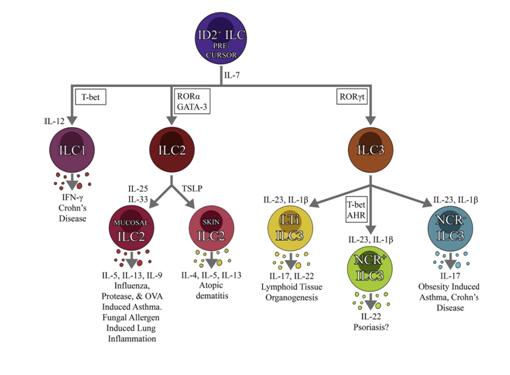
图3.各类ILC的发育和生物学功能
三)ILC2
ILC2于2001年以非T、非B细胞首次报导,在IL-25、IL-33和TSLP的刺激下增殖并释放IL-4、IL-5、IL-9和IL-13等II型细胞因子(图3)。小鼠ILC2表达CD25(IL-2受体)、CD90(Thy 1)、CD117(c-kit)和CD127(IL-7受体α链)等表面标志。此外,还表达可变数量的CD278(ICOS)、ST2(IL-33受体)和IL-17RB。ILC2分化发育受IL-7、IL-33、DNA结合抑制因子2(ID2)、RORα、GATA3和notch信号调控(图3)。
1.与早发型过敏性哮喘发病的关系:ILC2首先在肠道发现,提示其抵抗寄生虫感染的作用,后来证实在多种器官组织,包括人肺中也存在[7];在哮喘患者的外周血也存在,其比例明显高于正常人和过敏性鼻炎患者[8、9]。气道上皮细胞与某些微生物和寄生虫及其产物、过敏原接触,或受到某些物理损伤后释放TSLP、IL-33、IL-25,募集并激活ILC2,分泌IL-5和IL-13,引发独立于适应性免疫的免疫反应[10、11],表现为气道嗜酸性细胞炎症、气道高分泌和气道高反应,这些过敏性哮喘常见的病理改变。后来多个研究团队在RAG缺陷小鼠——适应性免疫缺乏鼠的实验中也观察到这一现象。近来的研究还显示肺内ILC2还分泌精氨酸酶1,它是急慢性过敏性哮喘发病中的一种关键酶(图1)[12]。尽管至今尚无ILC2在人类哮喘发病中的直接证据,但这些结果可以推测:在缺乏T、B细胞的情况下,ILC2足以诱发哮喘样症状,在人类哮喘发病中可能起重要作用。此外,有研究证实ILC2也表达MHCII分子,使其具有向CD4+T细胞提呈抗原的能力,并通过共刺激OX40L、IL-4和经接触依赖性机制促进Th2分化[13]。
2.与伴有鼻息肉的迟发型哮喘发病的关系:这一表型的特点:在12岁以后发作,伴有外周血嗜酸性粒细胞增多、鼻息肉,有时伴随阿斯匹林过敏;过敏原皮试常呈阳性反应,尽管患者缺乏过敏表现。MjÖsberg等[9]发现在慢性鼻-鼻窦炎(CRSwNP)患者的鼻息肉内存在ILC2。此后,其他学者也证实在这些患者的窦粘膜中ILC2的比例明显高于无鼻息肉的慢性鼻-鼻窦炎患者[14-16]。在CRSwNP的窦粘膜上皮中IL-25、IL-33和eotaxin-3含量也显著升高。此外,这些病人的鼻息肉组织内IL-5和IL-13的mRNA表达上调[14],外周血和组织内嗜酸性粒细胞增多[16]。
3.与病毒相关性哮喘和AHR形成的关系:2岁以后病毒感染可引起哮喘,也可诱发原有哮喘急性加重[17-19]。给小鼠鼻滴流感病毒A后能迅速诱发AHR,这一作用是通过对ILC2的刺激,并非依赖适应性免疫系统[20]。在病毒感染期间,肺泡巨噬细胞、上皮细胞和NKT细胞释放IL-33,激活ILC2,使之分泌II型细胞因子IL-5、IL-13,在病毒被清除后,仍持续分泌,导致持久性气道嗜酸性粒细胞炎症和AHR的临床表现[20、21]。
然而,也有相反的实验结果表明ILC2在诱发AHR的同时也通过释放双调蛋白改善受损肺组织的修复。因此,气道损伤与修复间的平衡影响着ILC2引起哮喘可能性。
三)ILC3
ILC3的分化、发育受转录因子ROEγt和GATA3调控,分三种亚型(图3),即:(1)淋巴样组织诱导细胞(LTi),是淋巴或淋巴样器官发育所必须,能分泌IL-17和IL-22;(2)产IL-22型ILC3,在皮肤、肺和肠道执行防御功能;(3)产IL-17型ILC3,表达CCR6,在某些结肠炎患者肠道内被激活,在哮喘动物模型的肺内也有活化[22]。
最近研究证实:以产IL-17型 ILC3的细胞因子或直接运用重组IL-17能使模型动物发生气道炎症,并通过平滑肌收缩产生AHR,因此推测IL-17可能在气道疾病,尤其是非过敏性哮喘的发病过程中起作用[23、24]。
肥胖是哮喘发生的主要危险因素,尤其是重症顽固性、激素抵抗型哮喘,其机制不清,但肯定与过敏性哮喘不同。
临床观察到一些肥胖者出现哮喘症状伴有血清IL-17水平的升高。Kim 等[25]以高脂饮食喂养小鼠致其肥胖后出现AHR,其肺内产IL-17型细胞明显增多,证实此种细胞是Lin-Thy1.2+scal-1+CCR6+ILC3。肥胖型哮喘不依赖适应性免疫系统,因为Rag-/-小鼠在喂养高脂食物肥胖后肺内ILC3增多,并产生AHR。IL-1β能刺激ILC3增殖,研究发现以产IL-17型ILC3过继输入Rag-/-I12rg-/-小鼠体内(缺乏适应性免疫和ILC细胞)后,小鼠恢复IL-1β诱导AHR作用。提示产IL-17型ILC3细胞本生即能介导AHR的产生。给IL-17-/-小鼠高脂喂养,即便肥胖后也不发生AHR,提示IL-17对AHR形成的重要影响。肥胖小鼠肺巨噬细胞分泌活性型IL-1β增多,并诱导产IL-17ILC3的增殖,以anakinra(IL-1R拮抗剂)短时处理阻断IL-1信号能阻止肥胖小鼠发生AHR,并大大减少肺内产IL-17型ILC3数量。更为重要的是在一组重症哮喘患者的支气管肺泡灌洗液存在产IL-17的ILC3,这提示ILC3不仅存在于哮喘患者中,而且可能对发病发挥重要作用。
四、小结
从过敏性或非过敏性哮喘动物模型和体外研究获取的信息改变了我们对适应性免疫和固有免疫调节原理的认识,并已展示出固有免疫在哮喘发生中的重要影响,在有些情况下固有免疫尤其是ILC可能与适应性免疫一样重要。尽管固有免疫细胞的表面标志和细胞内标志、与其他炎性细胞的相互作用还未完全阐明,其生物学功能还有待研究,但对于固有免疫的研究已经开创了一个崭新的领域,它有可能导致哮喘和其他过敏性疾病治疗的改善。
参考文献
1.Hammad, H. et al. Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite a llergen. J. Exp. Med. 207, 2097–2111 (2010).
2.Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
3.Willart, M.A. & Lambrecht, B.N. The danger within: endogenous danger signals, atopy and asthma. Clin. Exp. Allergy 39, 12–19 (2009).
4.Lam, D. et al. Airway house dust extract exposures modify allergen-induced airway hypersensitivity responses by TLR4-dependent and independent pathways. J. Immunol. 181, 2925–2932 (2008).
5.Albacker LA, et al. Invariant natural killer T cells recognize a fungal glycosphingolipid that can induce airway hyperreactivity. Nat Med 2013;19:1297–1304
6.Pichavant M, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med 2008;205:385–393.
7.Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical
source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012;36(3):451–463.
8.Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immuneresponse in peripheral blood from patients with asthma. J Allergy ClinImmunol. 2014;134(3):671–678。
9.Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62.
10.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20(6):791–800.
11.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188(3):1503–1513.
12.Bando JK, Nussbaum JC, Liang HE, Locksley RM. Type 2 innate lymphoid cells constitutively express arginase-I in the naive and inflamed lung. J Leukoc Biol. 2013;94(5):877–84.
13.Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol. 2014;192(5):2442–2448.
14.Miljkovic D, Bassiouni A, Cooksley C, Ou J, Hauben E, Wormald PJ, et al. Association between group 2 innate lymphoid cells enrichment, nasal polyps and allergy in chronic rhinosinusitis. Allergy. 2014;69(9):1154–61.
15.Shaw JL, Fakhri S, Citardi MJ, Porter PC, Corry DB, Kheradmand F, et al IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am J Respir Crit Care Med. 2013;188(4):432–9.
16.Ho J, Bailey M, Zaunders J, Mrad N, Sacks R, Sewell W, et al. Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin Exp Allergy. 2015;45(2):394–403.
17.Singh AM, Moore PE, Gern JE, Lemanske Jr RF, Hartert TV. Bronchiolitis to asthma: a review and call for studies of gene-virus interactions in asthma causation. Am J Respir Crit Care Med. 2007;175(2):108–19.
18.Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol. 2010;84(15):7418–26.
19.Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Lee WM, et al. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am J Respir Crit Care Med. 2012;185(3):281–5.
20.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8.
21.Gorski SA, Hahn YS, Braciale TJ. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. 2013;9(9), e1003615. doi:10.1371/journal.ppat.1003615.
22.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al.Interleukin- 17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 2014;20:54-61.
23.Kudo, M., A. C. Melton, C. Chen, M. B. Engler, K. E. Huang, X. Ren, Y. Wang, X. Bernstein, J. T. Li, K. Atabai, X. Huang, and D. Sheppard. 2012. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Med. 18: 547-554.
24.McKinley, L., J. F. Alcorn, A. Peterson, R. B. Dupont, S. Kapadia, A. Logar, A. Henry, C. G. Irvin, J. D. Piganelli, A. Ray, and J. K. Kolls. 2008. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 181: 4089-4097.
25.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate上obesity-associated airway hyperreactivity. Nat Med 2014;20:54-61.
ILC2于2001年以非T、非B细胞首次报导,在IL-25、IL-33和TSLP的刺激下增殖并释放IL-4、IL-5、IL-9和IL-13等II型细胞因子(图3)。小鼠ILC2表达CD25(IL-2受体)、CD90(Thy 1)、CD117(c-kit)和CD127(IL-7受体α链)等表面标志。此外,还表达可变数量的CD278(ICOS)、ST2(IL-33受体)和IL-17RB。ILC2分化发育受IL-7、IL-33、DNA结合抑制因子2(ID2)、RORα、GATA3和notch信号调控(图3)。
1.与早发型过敏性哮喘发病的关系:ILC2首先在肠道发现,提示其抵抗寄生虫感染的作用,后来证实在多种器官组织,包括人肺中也存在[7];在哮喘患者的外周血也存在,其比例明显高于正常人和过敏性鼻炎患者[8、9]。气道上皮细胞与某些微生物和寄生虫及其产物、过敏原接触,或受到某些物理损伤后释放TSLP、IL-33、IL-25,募集并激活ILC2,分泌IL-5和IL-13,引发独立于适应性免疫的免疫反应[10、11],表现为气道嗜酸性细胞炎症、气道高分泌和气道高反应,这些过敏性哮喘常见的病理改变。后来多个研究团队在RAG缺陷小鼠——适应性免疫缺乏鼠的实验中也观察到这一现象。近来的研究还显示肺内ILC2还分泌精氨酸酶1,它是急慢性过敏性哮喘发病中的一种关键酶(图1)[12]。尽管至今尚无ILC2在人类哮喘发病中的直接证据,但这些结果可以推测:在缺乏T、B细胞的情况下,ILC2足以诱发哮喘样症状,在人类哮喘发病中可能起重要作用。此外,有研究证实ILC2也表达MHCII分子,使其具有向CD4+T细胞提呈抗原的能力,并通过共刺激OX40L、IL-4和经接触依赖性机制促进Th2分化[13]。
2.与伴有鼻息肉的迟发型哮喘发病的关系:这一表型的特点:在12岁以后发作,伴有外周血嗜酸性粒细胞增多、鼻息肉,有时伴随阿斯匹林过敏;过敏原皮试常呈阳性反应,尽管患者缺乏过敏表现。MjÖsberg等[9]发现在慢性鼻-鼻窦炎(CRSwNP)患者的鼻息肉内存在ILC2。此后,其他学者也证实在这些患者的窦粘膜中ILC2的比例明显高于无鼻息肉的慢性鼻-鼻窦炎患者[14-16]。在CRSwNP的窦粘膜上皮中IL-25、IL-33和eotaxin-3含量也显著升高。此外,这些病人的鼻息肉组织内IL-5和IL-13的mRNA表达上调[14],外周血和组织内嗜酸性粒细胞增多[16]。
3.与病毒相关性哮喘和AHR形成的关系:2岁以后病毒感染可引起哮喘,也可诱发原有哮喘急性加重[17-19]。给小鼠鼻滴流感病毒A后能迅速诱发AHR,这一作用是通过对ILC2的刺激,并非依赖适应性免疫系统[20]。在病毒感染期间,肺泡巨噬细胞、上皮细胞和NKT细胞释放IL-33,激活ILC2,使之分泌II型细胞因子IL-5、IL-13,在病毒被清除后,仍持续分泌,导致持久性气道嗜酸性粒细胞炎症和AHR的临床表现[20、21]。
然而,也有相反的实验结果表明ILC2在诱发AHR的同时也通过释放双调蛋白改善受损肺组织的修复。因此,气道损伤与修复间的平衡影响着ILC2引起哮喘可能性。
三)ILC3
ILC3的分化、发育受转录因子ROEγt和GATA3调控,分三种亚型(图3),即:(1)淋巴样组织诱导细胞(LTi),是淋巴或淋巴样器官发育所必须,能分泌IL-17和IL-22;(2)产IL-22型ILC3,在皮肤、肺和肠道执行防御功能;(3)产IL-17型ILC3,表达CCR6,在某些结肠炎患者肠道内被激活,在哮喘动物模型的肺内也有活化[22]。
最近研究证实:以产IL-17型 ILC3的细胞因子或直接运用重组IL-17能使模型动物发生气道炎症,并通过平滑肌收缩产生AHR,因此推测IL-17可能在气道疾病,尤其是非过敏性哮喘的发病过程中起作用[23、24]。
肥胖是哮喘发生的主要危险因素,尤其是重症顽固性、激素抵抗型哮喘,其机制不清,但肯定与过敏性哮喘不同。
临床观察到一些肥胖者出现哮喘症状伴有血清IL-17水平的升高。Kim 等[25]以高脂饮食喂养小鼠致其肥胖后出现AHR,其肺内产IL-17型细胞明显增多,证实此种细胞是Lin-Thy1.2+scal-1+CCR6+ILC3。肥胖型哮喘不依赖适应性免疫系统,因为Rag-/-小鼠在喂养高脂食物肥胖后肺内ILC3增多,并产生AHR。IL-1β能刺激ILC3增殖,研究发现以产IL-17型ILC3过继输入Rag-/-I12rg-/-小鼠体内(缺乏适应性免疫和ILC细胞)后,小鼠恢复IL-1β诱导AHR作用。提示产IL-17型ILC3细胞本生即能介导AHR的产生。给IL-17-/-小鼠高脂喂养,即便肥胖后也不发生AHR,提示IL-17对AHR形成的重要影响。肥胖小鼠肺巨噬细胞分泌活性型IL-1β增多,并诱导产IL-17ILC3的增殖,以anakinra(IL-1R拮抗剂)短时处理阻断IL-1信号能阻止肥胖小鼠发生AHR,并大大减少肺内产IL-17型ILC3数量。更为重要的是在一组重症哮喘患者的支气管肺泡灌洗液存在产IL-17的ILC3,这提示ILC3不仅存在于哮喘患者中,而且可能对发病发挥重要作用。
四、小结
从过敏性或非过敏性哮喘动物模型和体外研究获取的信息改变了我们对适应性免疫和固有免疫调节原理的认识,并已展示出固有免疫在哮喘发生中的重要影响,在有些情况下固有免疫尤其是ILC可能与适应性免疫一样重要。尽管固有免疫细胞的表面标志和细胞内标志、与其他炎性细胞的相互作用还未完全阐明,其生物学功能还有待研究,但对于固有免疫的研究已经开创了一个崭新的领域,它有可能导致哮喘和其他过敏性疾病治疗的改善。
参考文献
1.Hammad, H. et al. Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite a llergen. J. Exp. Med. 207, 2097–2111 (2010).
2.Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
3.Willart, M.A. & Lambrecht, B.N. The danger within: endogenous danger signals, atopy and asthma. Clin. Exp. Allergy 39, 12–19 (2009).
4.Lam, D. et al. Airway house dust extract exposures modify allergen-induced airway hypersensitivity responses by TLR4-dependent and independent pathways. J. Immunol. 181, 2925–2932 (2008).
5.Albacker LA, et al. Invariant natural killer T cells recognize a fungal glycosphingolipid that can induce airway hyperreactivity. Nat Med 2013;19:1297–1304
6.Pichavant M, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med 2008;205:385–393.
7.Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical
source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012;36(3):451–463.
8.Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immuneresponse in peripheral blood from patients with asthma. J Allergy ClinImmunol. 2014;134(3):671–678。
9.Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62.
10.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20(6):791–800.
11.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188(3):1503–1513.
12.Bando JK, Nussbaum JC, Liang HE, Locksley RM. Type 2 innate lymphoid cells constitutively express arginase-I in the naive and inflamed lung. J Leukoc Biol. 2013;94(5):877–84.
13.Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol. 2014;192(5):2442–2448.
14.Miljkovic D, Bassiouni A, Cooksley C, Ou J, Hauben E, Wormald PJ, et al. Association between group 2 innate lymphoid cells enrichment, nasal polyps and allergy in chronic rhinosinusitis. Allergy. 2014;69(9):1154–61.
15.Shaw JL, Fakhri S, Citardi MJ, Porter PC, Corry DB, Kheradmand F, et al IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am J Respir Crit Care Med. 2013;188(4):432–9.
16.Ho J, Bailey M, Zaunders J, Mrad N, Sacks R, Sewell W, et al. Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin Exp Allergy. 2015;45(2):394–403.
17.Singh AM, Moore PE, Gern JE, Lemanske Jr RF, Hartert TV. Bronchiolitis to asthma: a review and call for studies of gene-virus interactions in asthma causation. Am J Respir Crit Care Med. 2007;175(2):108–19.
18.Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol. 2010;84(15):7418–26.
19.Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Lee WM, et al. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am J Respir Crit Care Med. 2012;185(3):281–5.
20.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8.
21.Gorski SA, Hahn YS, Braciale TJ. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. 2013;9(9), e1003615. doi:10.1371/journal.ppat.1003615.
22.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al.Interleukin- 17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 2014;20:54-61.
23.Kudo, M., A. C. Melton, C. Chen, M. B. Engler, K. E. Huang, X. Ren, Y. Wang, X. Bernstein, J. T. Li, K. Atabai, X. Huang, and D. Sheppard. 2012. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Med. 18: 547-554.
24.McKinley, L., J. F. Alcorn, A. Peterson, R. B. Dupont, S. Kapadia, A. Logar, A. Henry, C. G. Irvin, J. D. Piganelli, A. Ray, and J. K. Kolls. 2008. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 181: 4089-4097.
25.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate上obesity-associated airway hyperreactivity. Nat Med 2014;20:54-61.
上一篇:
重症哮喘的诊断、评估和个体化治疗
下一篇:
重症嗜酸性粒细胞型哮喘









